RNAseq Read Count Analysis¶
Analyzing Read Count¶
According to this tool, it is possible to know exactly how many reads matched a given genomic object of the reference sequence. Results are accessible following a 5 steps process which is described below.

1. Choose one or several reference sequences.
2. Select at least one experiment and compute the associated read count number per genomic object. (check publication for terminology of experiments, which is displayed in the head of the interface: Sharma et al, 2010, Nature 464:250-255 for the given example)
3. It is possible to restrict the query to one or several given classes of genomic objects ( CDS, fCDS, rRNA, tRNA, miscRNA or all ).
4. Query can be constrained upon the strand of the transcripts (direct, reverse, both)
5. Submit query.
As usual, results are reported in a table which is composed of 3 main sections (see below).
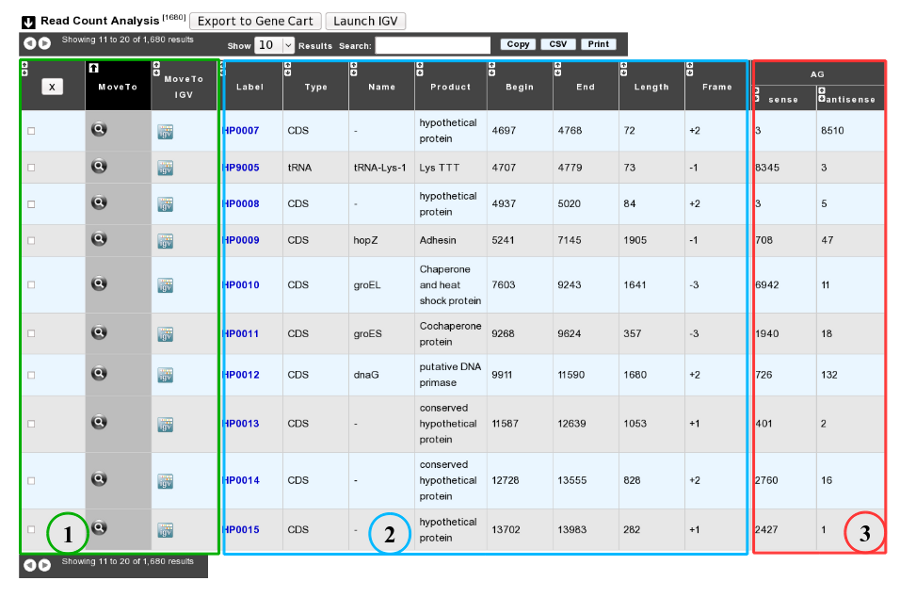
1. Export functions. This section allows users to make all genes (or subsets of genes) available for other analysis tools. 3 main operations are possible here:
select subsets of genes (by selecting checkboxes on the first column) and export them into a Gene Cart by using the “Export To Gene Cart” button.
See one selected gene into the MaGe Genome Browser by clicking on the magnifying glass.
Direct link to the selected gene in Integrative Genome Viewer.
2. The second part reports the main genomic object features : Label (Link to more Genomic Object information), Type, Name, Product, Begin, End, Length, Frame.
3. RNA-Seq Result part : Read count (direct and/or reverse)