Gene annotation editor
Overview of the annotation editor
How to access to the Gene Annotation Editor?
There are two ways of accessing the Gene Annotation Editor:
click on a genomic object on the genomic map
click on a label in the table of genomic objects which is below the genomic map
Important
requesting information via the GetInfo button only calls up a read-only Gene Annotation Editor window.
Overview of the Gene Annotation Editor
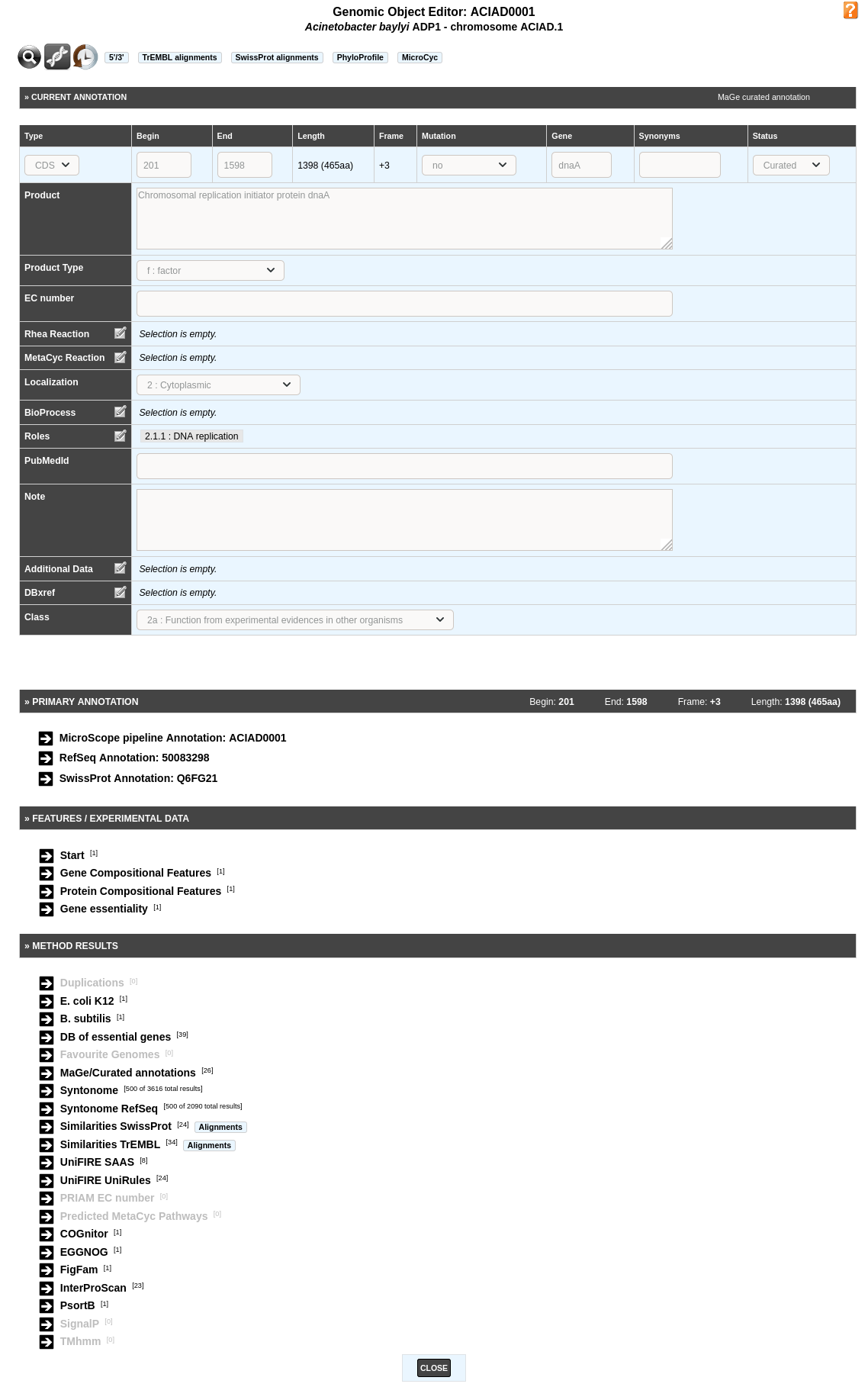
The Gene Annotation Editor window is made of 5 sections:
a toolbar that allows access to different functionalities
the current annotation of the genomic object. This section can be modified by the annotator (with sufficient rights).
the primary annotation of the genomic object. It corresponds to the MicroScope pipeline automatic annotation (if it is a first annotation) or to the databank annotation (if it is a reannotation project).
the Feature / Experimental data section. This section provides access to some features of the gene (i.e. star codon prediction, nucleic or amino-acid composition) and to experimental data if any (e.g. gene essentiality, transposon mutants, SNPs/InDels).
the Method results section. This section gives an access to the results obtained by the different tools for comparative and functional analyses.
How to use the Gene Annotation Editor toolbar?

It contains several buttons allowing access to different functionalities:
the first button allows to open the genomic object in the viewer
the second button allows to access the sequence (nucleic and protein) of the genomic object
the third button allows to access the annotation history of the genomic object
5’/3’: the nucleic sequence of the genomic object + the nucleic context
TrEMBL alignments: visualisation of the alignments with TrEMBL best hits
SwissProt alignments: visualisation of the alignments with SwissProt best hits
Phyloprofile: this tool provides a list of all CDSs (from all replicons) that have the same phylogenetic profile (presence/absence of homologue in others species) than the current genomic object. Note: query can be slow.
PubMed: this functionality opens a new window that shows the references that have been linked to this genomic object on PubMed (this button is not displayed if no reference are linked to this Genomic Object)
KEGG: this functionality opens the KEGG description corresponding to the annotated EC number(s)
Brenda: this functionality opens the Brenda entry corresponding to the annotated EC number(s)
MicroCyc: this functionality opens a new window showing information related to the genomic object in the MicroCyc database
Expert annotation of gene function
How to fill the Gene Annotation form?
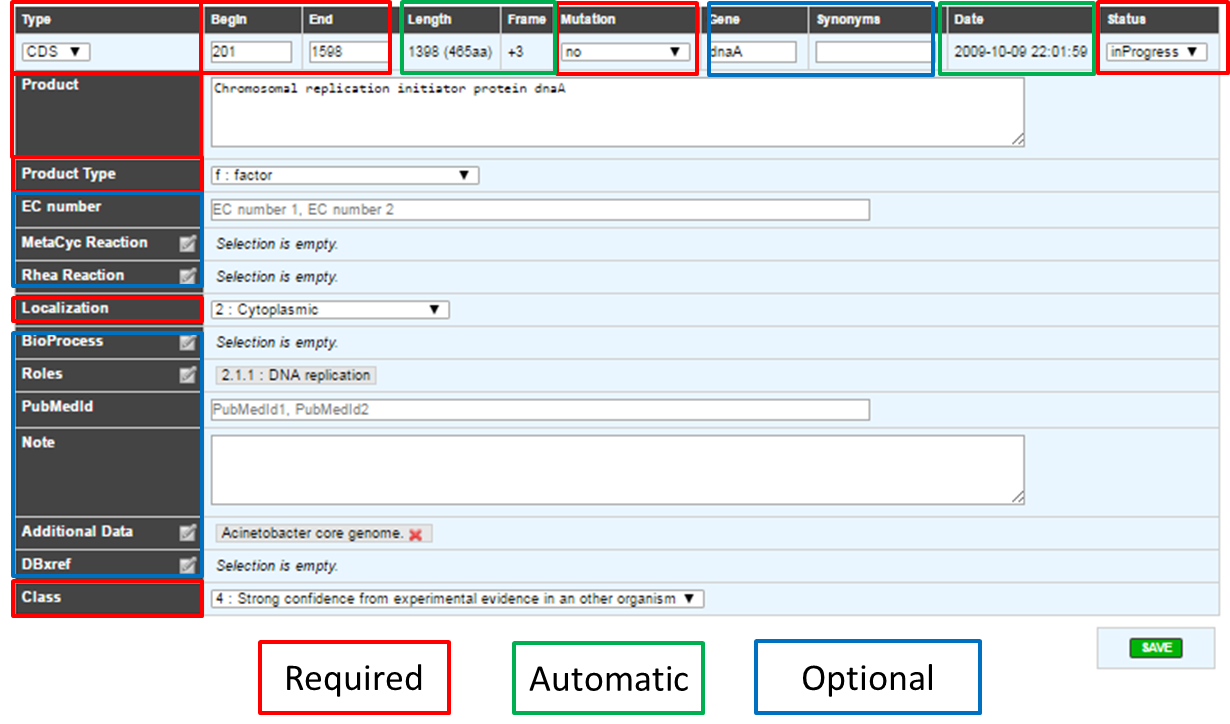
As shown in the figure below, not all fields can be modified by the annotator. Furthermore, some of them are required and other are optional. These fields have to be filled after the careful analysis of the different methods results. If your are working on other object than CDS, you may have a different form, if a required field for CDS appear in your form, it’s still required.
Tip
If one of the required field is missing or wrongly filled a warning will appear in the window.
What are the different annotation “Status”?
inProgress : the annotator has not finished the expert annotation
finished : the annotator has finished the expert annotation
Curated : the expert annotation has been reviewed by a specialist of the functional process in which the CDS product is involved
Artefact : An artefactual CDS corresponds to a false prediction by the gene detection program. An artefactual CDS should never be similar to any proteins from the databanks (except if the same erroneous annotation has been made in another genomes)
chkSeq : this status is used by the annotator to flag potential sequencing errors in the sequence. When the sequencing is performed at Genoscope, these chkSeq sequences will be sent to the people working in the finishing team. They will then check the assembly to see if the sequence quality is good or not. If needed they can perform some additional PCRs to enhance the data.
chkStart : the annotator suspects that a start position readjustment might be needed for the CDS, but hasn’t done it yet.
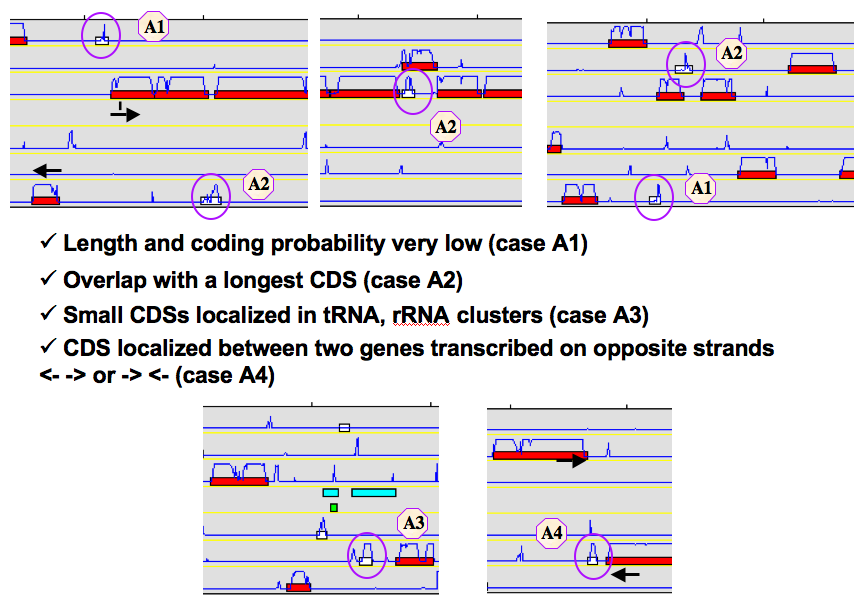
How to identify artefacts?
What are the different “Type” categories?
CDS
fCDS
tRNA
rRNA
misc_RNA
tmRNA
ncRNA
IS
misc_feature
promoter
How to fill the “Mutation” field?
no => Normal CDS
frameshift => CDS for which a true frame-shift has been biologically demonstrated
pseudo => the CDS is part of a pseudogene
partial => the CDS is a gene fragment
gene remnant => the CDS is a highly degraded gene fragment
selenocysteine => the CDS contains a Selenocysteine in its sequence
pyrrolysine => the CDS contains a pyrrolysine in its sequence
What are the different “Product type” categories?
u : unknown
n : RNA
e : enzyme
f : factor
r : regulator
c : carrier
t : transporter
rc : receptor
s : structure
l : leader peptide
m : membrane component
lp : lipoprotein
cp : cell process
ph : phenotype
h : extrachromosomal origin
How to use the “MetaCyc reaction” field?
This field allows user to link one ore more metabolic reactions from MetaCyc (BioCyc) to the current edited gene.
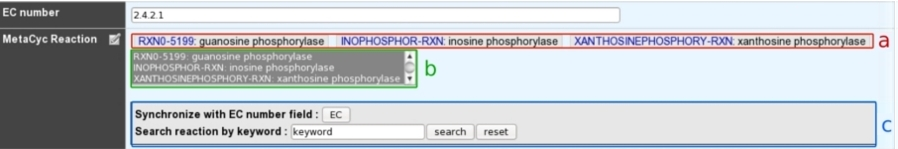
a: Reactions presented at the top of the field have been manually curated by an annotator.
b: A multiple selection list gives quick access to all predicted (unselected) or curated (selected) reactions linked to this gene.
c: A search box allows one to quickly access MetaCyc reactions corresponding to either EC numbers from previous EC number field or a given keyword.
Search box :
Clicking on the “EC” button will search all MetaCyc reactions corresponding to the EC number from the “EC number” field.
The keyword search will look for all MetaCyc reactions having an identifier, a name or involving a compound similar to the given keyword.
Search result :

The search returns a list of MetaCyc reactions, with :
the reaction identifier and name. Identifier is clickable and open the BioCyc reaction card.
And in some cases :
Genes of the organism already linked to this reaction (eg. first row of the example). Genes are flagged with :
“validated” : reaction has been manually linked to this gene by users.
“annotated” : reaction has been linked to homologous gene and transferred here from a close genome.
“predicted” : reaction has been linked to this gene by the pathway-tools algorithm.
If the reaction has no known coding genes but belongs to a pathway predicted to exist in the current organism, a clickable link to the MetaCyc pathway description is given (eg. fourth row of the example).
The “Reset” button deletes all results.
How to use the “Rhea reaction” field?
This field allows user to link one ore more metabolic reactions from Rhea to the current edited gene.
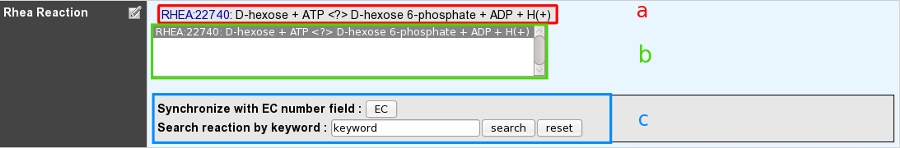
a: Reactions presented at the top of the field have been manually curated by an annotator.
b: A multiple selection list gives quick access to all curated reactions linked to this gene.
c: A search box allows one to quickly access Rhea reactions corresponding to either EC numbers from previous EC number field or a given keyword.
Search box :
Clicking on the “EC” button will search all Rhea reactions corresponding to the EC number from the “EC number” field.
The keyword search will look for all Rhea reactions having an identifier, a name, involving a compound name or Chebi identifier similar to the given keyword.
Search result :
Rhea reactions are present in 4 exemplary according to the direction :
bidirectional : <=>
left to right : =>
right to left : <=
*unknown (master reaction) : <?>
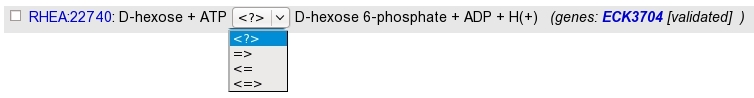
The search returns a list of Rhea reactions, with :
the reaction identifier and name. Identifier is clickable and open the Rhea reaction card. By default, the master reaction is presented. Select the direction wanted in the “direction-select”.
And in some cases :
Genes of the organism already linked to this reaction (eg. first row of the example). Genes are flagged with :
“validated” : reaction has been manually linked to this gene by users.
The “Reset” button deletes all results
How to link a new reaction :
For each reaction in the result set, check-box allows to add a reaction from the result set to the selected element. All reactions selected in the multiple selection list will be saved as validated and linked to this gene. Unselecting a reaction in this list will remove this link from the curated data.
What are the different “Localization” categories?
1 : Unknown
2 : Cytoplasmic
3 : Fimbrial
4 : Flagellar
5 : Inner membrane protein
6 : Inner membrane-associated
7 : Outer membrane protein
8 : Outer membrane-associated
9 : Periplasmic
10 : Secreted
11 : Membrane
What is the “BioProcess” classification?
This functional classification is based on the CMR JCVI Role IDs.
This field is optionally filled in during the expert annotation process.
What is the “Roles” classification?
This functional classification corresponds to the MultiFun classification which has been developed by Monica Riley for E. coli.
This field is optionally filled in during the expert annotation process.
How to use the “PubMedID” field?
The PubMedID or PMID correspond to the index of a publication on the PubMed section of the NCBI website. You can fill this field when you want to link a publication to your annotation. If you want to enter several publications, you simply have to write the PMIDs separated by commas.
You will find the PMID of a publication directly on Pubmed as shown on the figure below. You can also find PMIDs in the “References” section of the UniProt entries.
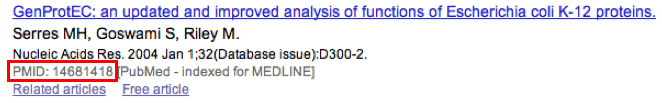
If this field is filled you will have a direct access to the publications on PubMed by clicking on the PubMed button on top of the Gene annotation editor window.
How to use the “Additional data” field?
The Comments field is dedicated to the annotators who want to leave some notes for themselves or for others annotators.
How to use the “Class” field?
The Class annotation categories are useful for assigning a “confidence level” to each gene annotation. It has been inspired by the “protein name confidence” defined in PseudoCAP (Pseudomonas aeruginosa community annotation project).
This information is not given by the automatic functional annotation procedure, except in case of functional annotation transfer from a genome being annotated with MaGe.
The different classes are:
1a : Function from experimental evidences in the studied strain
1b : Function from experimental evidences in the studied species
1c : Function from experimental evidences in the studied genus
2a : Function from experimental evidences in other organisms
2b : Function from indirect experimental evidences (e.g. phenotypes)
3 : Putative function from multiple computational evidences
4 : Unknown function but conserved in other organisms
5 : Unknown function
How to choose the “Class” annotation category?
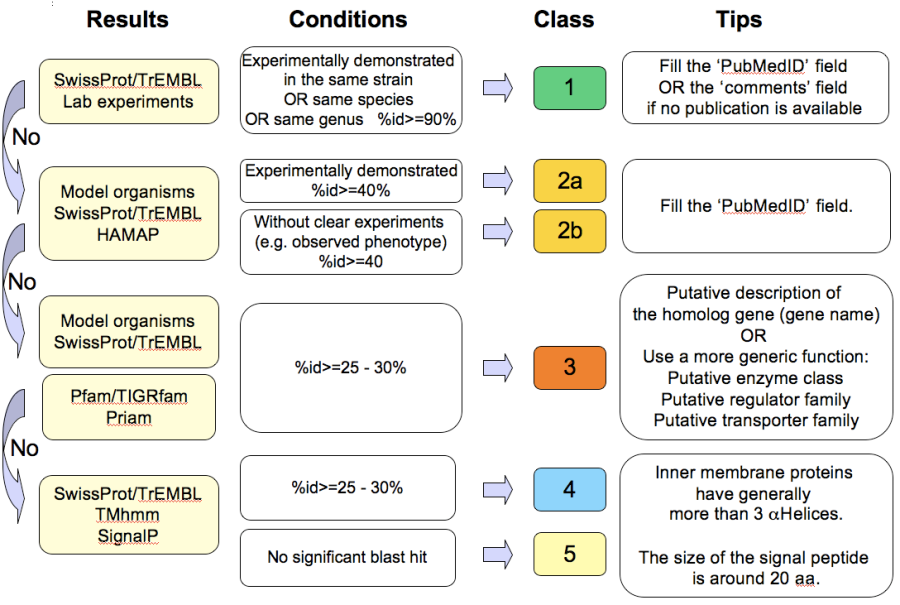
Annotation Rules
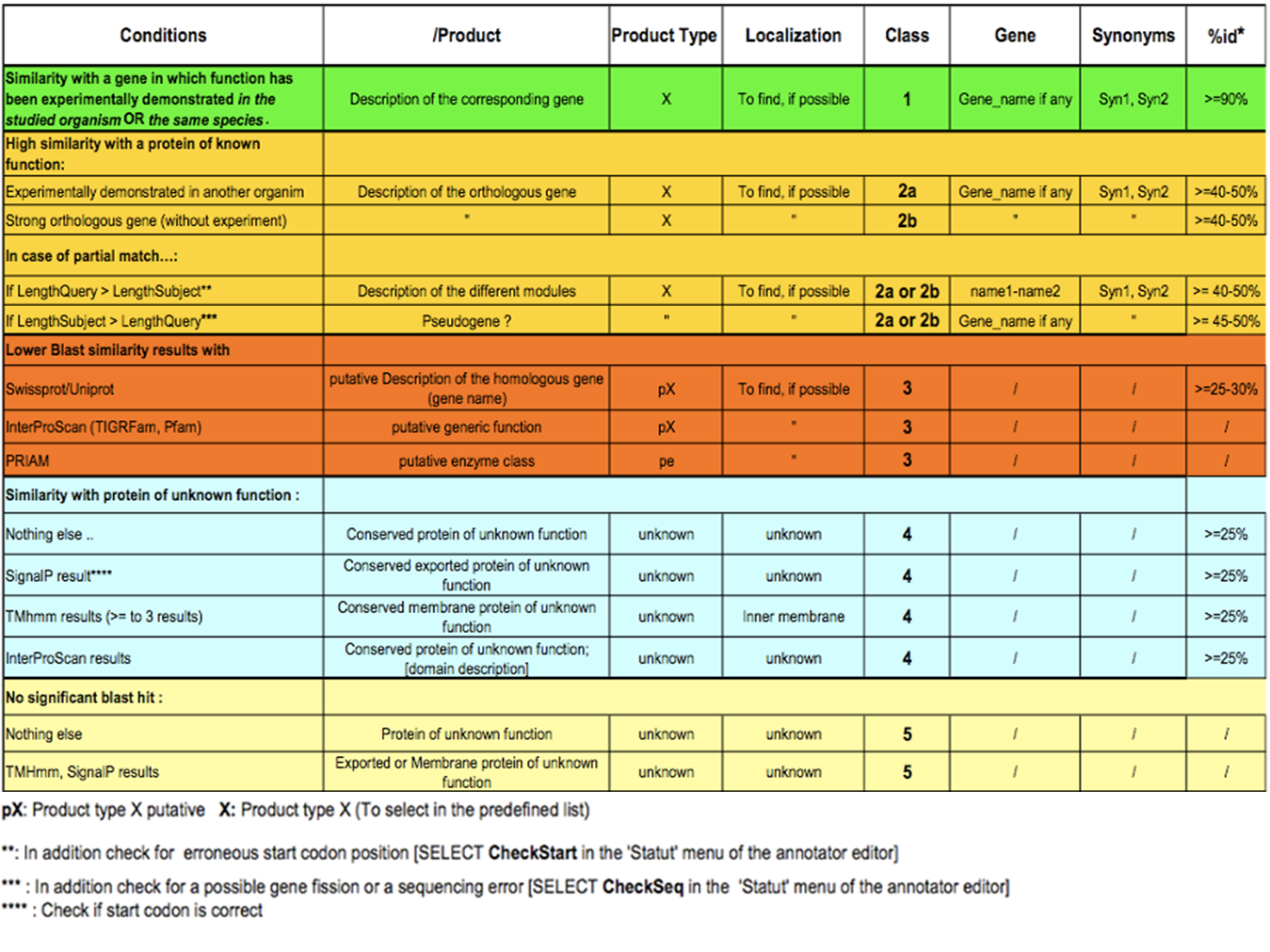
Considering the Class field, here are some basic annotation rules:
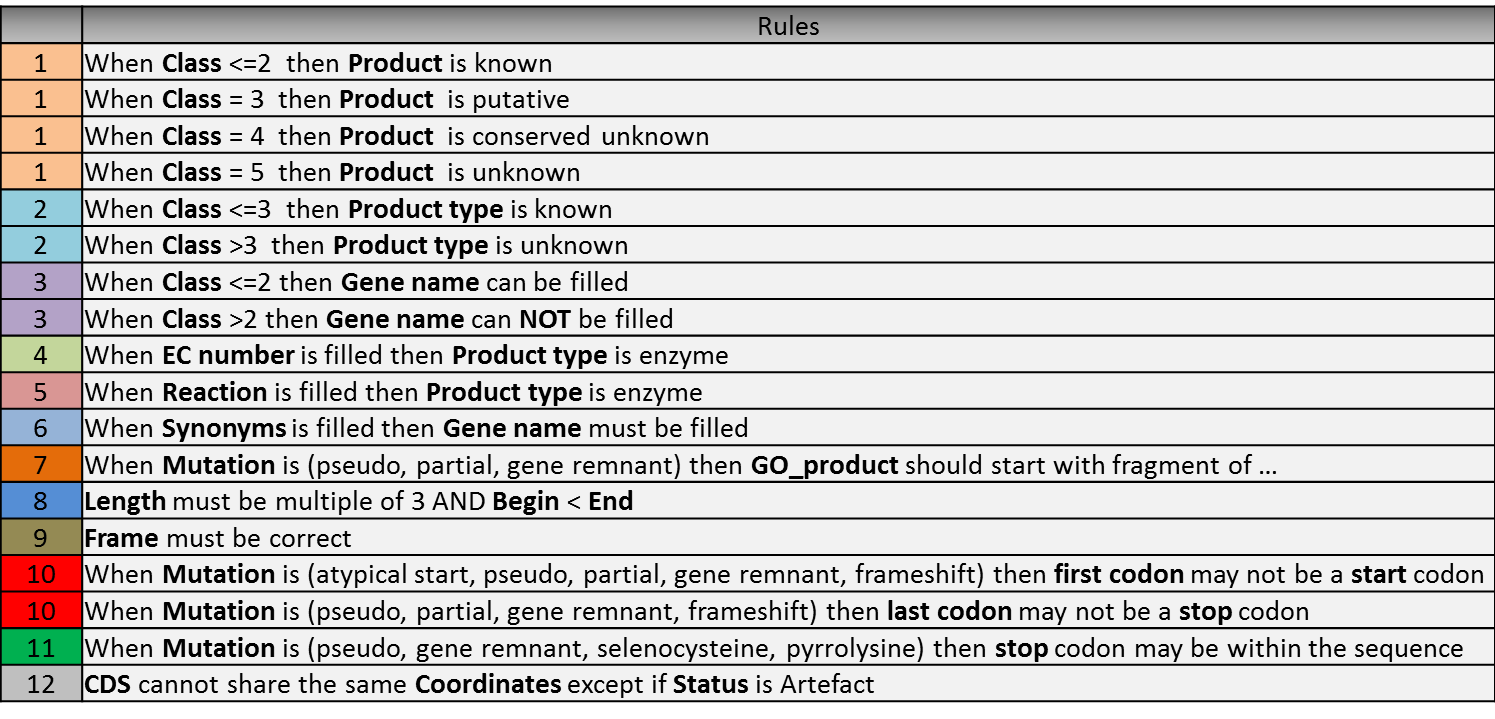
1 a/b/c: Function from experimental evidences in the studied organism/species/genus
Gene [optional]
Synonyms [optional]
Product [known]
EC number [optional]
MetaCyc Reaction [optional]
PubMedId [known]
ProductType [known]
Localization [optional]
BioProcess [optional]
Roles [optional]
2a : Function from experimental evidences in other organism
Gene [optional]
Synomyms [optional]
Product [known]
EC number [optional]
MetaCyc Reaction [optional]
PubMedId [known]
ProductType [known]
Localization [optional]
BioProcess [optional]
Roles [optional]
2b : Function from indirect experimental evidences (e.g. phenotypes)
Gene [optional]
Synonyms [optional]
Product [known]
EC number [optional]
MetaCyc Reaction [optional]
PubMedId [optional]
ProductType [known]
Localization [optional]
BioProcess [optional]
Roles [optional]
3 : Putative function from multiple computational evidences
Gene [not allowed]
Synonyms [not allowed]
Product [putative function]:
EC number [optional]
MetaCyc Reaction [optional]
PubMedId [optional]
ProductType [known]
Localization [optional]
BioProcess [optional]
Roles [optional]
4 : Unknown function but conserved in other organisms
Gene [not allowed]
Synonyms [not allowed]
Product [conserved … protein of unknown function … ]
EC number [not allowed]
MetaCyc Reaction [optional]
PubMedId [optional]
ProductType [u : unknown]
Localization [optional]
BioProcess [optional]
Roles [optional]
5 : Unknown function
Gene [not allowed]
Synonyms [not allowed]
Product [protein of unknown function]
EC number [not allowed]
MetaCyc Reaction [optional]
PubMedId [optional]
ProductType [u : unknown]
Localization [optional]
BioProcess [optional]
Roles [optional]
Start
In progress
This menu gives the beginning and the end of the gene sequence according to different softwares. If the indicated start and stops seems to be wrong when compared to those given by the softwares, you can correct them by using Artemis (see Artemis).
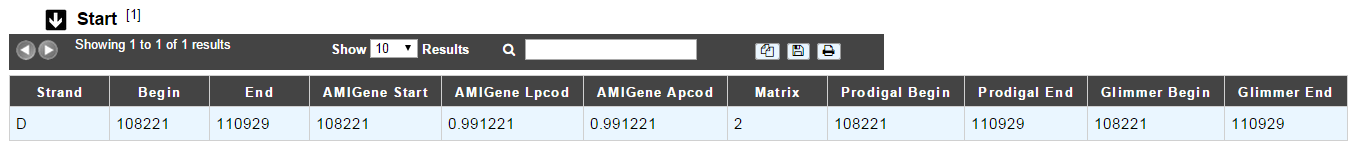
Strand: indicates if the CDS is on the direct strand (D) or on the reverse strand (R)
Begin: give the leftmost beginning of the CDS according to the expert or automatic annotations
End: give the ending of the CDS according to the expert or automatic annotations
AMIGene Start: gives the start according to AMIGene
AMIGene Lpcod: gives the coding probability on the length End-Begin +1 according to AMIGene
AMIGene Apcod: gives the length End-AMstart +1 according to AMIGene
Matrix: gives the matrix number (see here)
SHOW Begin: gives the position of the first nucelic acid of the CDS according to SHOW
SHOW End: gives the position of the last nucelic acid of the CDS according to SHOW
SHOW Proba : gives the coding probability on the lenght End-SHOW begin +1 according to SHOW
Prodigal Begin: give the beginning of the CDS according to the expert or automatic annotation
Prodigal End: give the ending of the CDS according to the expert or automatic annotation
Compositional features
Gene compositional features
This section gives the different compositional features of the studied gene, determined by GenProtFeat.
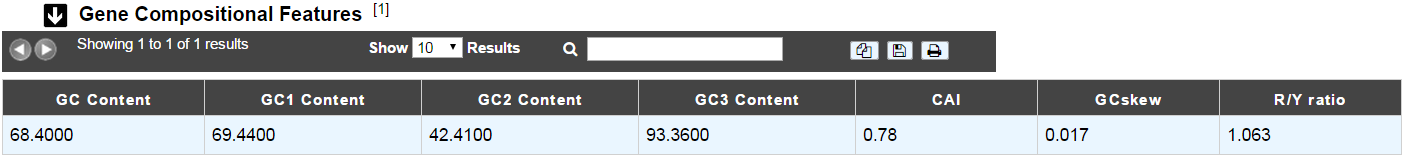
GC Content:
GC1 Content:
GC2 Content:
GC3 Content:
CAI:
GCskew:
R/Y ratio:
Protein compositional features
This section gives the different compositional features of the studied gene, determined by GenProtFeat.

Mw (Da): gives the molecular weight of the protein (Da)
Hydrophobicity:
Tiny:
Small:
Aliphatic:
Aromatic:
NonPolar:
Polar:
Charged:
Basic:
Acidic:
PI: gives the value of the protein isoelectric point
Oxyphobic Index:
Duplications
This dataset contains the list of genes of the genome that have an identity > 25% with a minLRap > 0.75 to the selected gene.
How to read the result table?
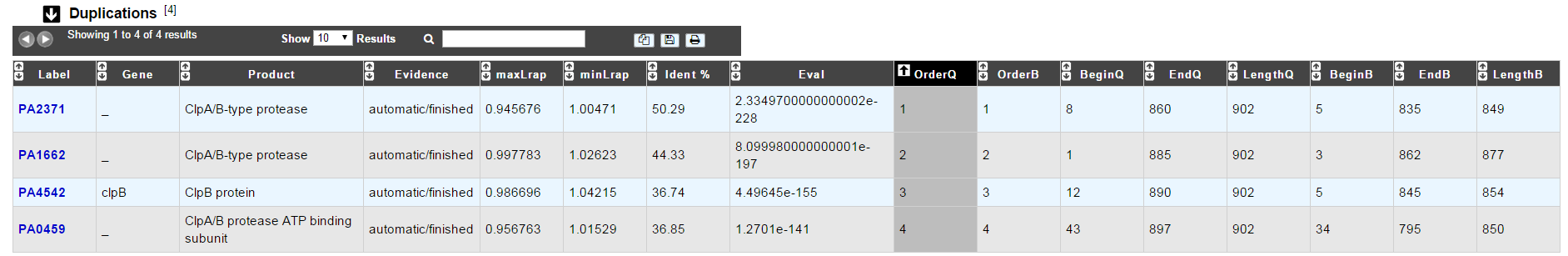
Label: Label of the protein. If you click on the label, you access to the Gene annotation window
Gene: Gene name of the protein
Product: Product description of the protein
maxLrap: see BLAST results
minLrap: see BLAST results
Ident%: Percentage of identity between the studied protein and the database protein
Eval: E value of the BLAST result
OrderQ: see BLAST results
OrderB: see BLAST results
BeginQ: Start of the alignment for the studied protein
EndQ: End of the alignment for the studied protein
LengthQ: Length of the studied protein
BeginB: Start of the alignment for the database protein
EndB: End of the alignment for the database protein
LengthB: Length of the database protein
E. coli K12
In progress
This menu indicates the best BLAST hit for the current Genomic Object against the genome of Escherichia coli K12, if any.
This dataset is a useful reference since E. coli is a very well known bacteria, with a carefully annotated genome and large quantities of experimental data and publications are available.
Tip
This dataset can help you to complete your expert annotation.
How to read the result table?
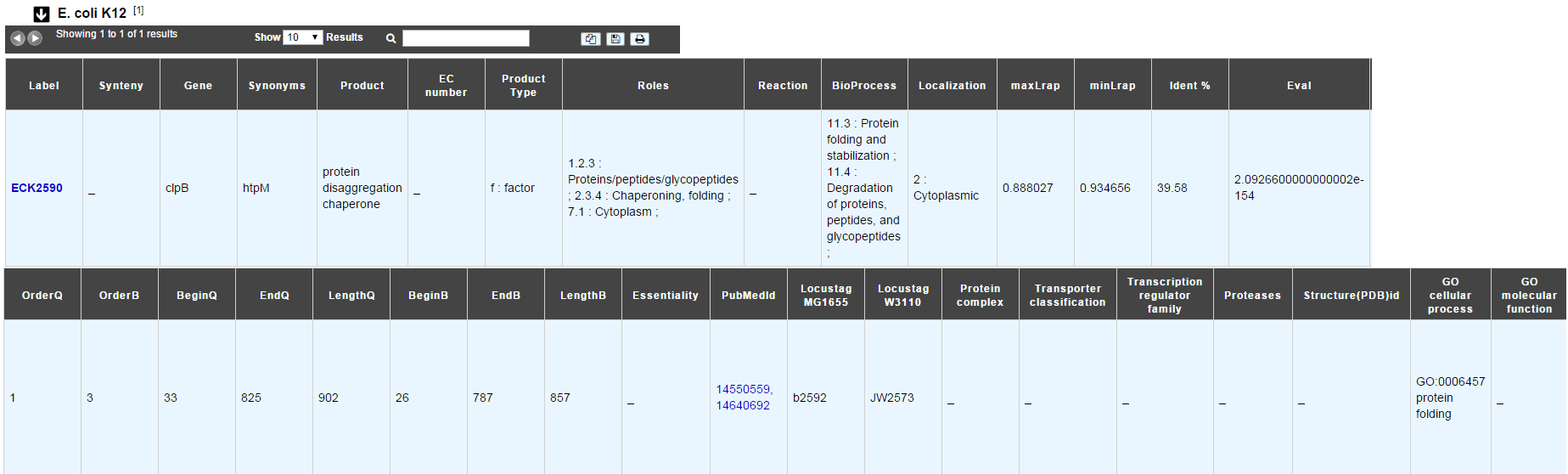
Label: Label of the protein. If you click on the label, you access to the Gene annotation window
Synteny: If you click on the magnifying glass, it opens a synton visualisation window (if any)
Gene: Gene name of the protein
Synonyms: Alternative name for the gene (if any)
Product: Product description of the protein
ECnumber: EC number associated with the protein, if any
Product type: Description of the product type of the protein
Roles: Functional categories associated with the protein using the Roles functional classification
Reaction: If any, gives the reactions implying the database protein (reactions given by Rhea and MetaCyc)
BioProcess: Functional categories associated with the protein using the BioProcess functional classification
Localization: Cellular localisation of the protein
maxLrap: see BLAST results
minLrap: see BLAST results
Ident%: Percentage of identity between the studied protein and the database protein
Eval: E value of the BLAST result
OrderQ: see BLAST results
OrderB: see BLAST results
BeginQ: Start of the alignment for the studied protein
EndQ: End of the alignment for the studied protein
LengthQ: Length of the studied protein
BeginB: Start of the alignment for the database protein
EndB: End of the alignment for the database protein
LengthB: Length of the database protein
PubMedId: PubMed references linked to the annotation of the protein
Locustag MG1655: locus tag of the gene in the regulon of LeuO in E coli K12 (??)
Locustag W3110: locus tag of the gene in the NarP regulon of E coli K12 (??)
Protein complex: Indicates if the database protein is part of a protein complex
Transporter classification: If the database protein is a transporter, indicates the family this transporter is part of
Transcription regulator family: If the database protein is a transcription regulator, indicates the family this transcription regulator is part of
Proteases: If the database protein is a protease, indicates the family this protease is part of
Structure(PDB)id: Gives the Id number which correspond to the database protein’s structure on Protein Data Bank
GO cellular process: Gives the cellular process according to Gene Ontology
GO molecular function: Gives the molecular process according to Gene Ontology
B. subtilis
This menu indicates the best BLAST hit for the current Genomic Object against the genome of Bacillus subtilis, if any.
This dataset is a useful reference since B. subtilis is a very well known bacteria, with a carefully annotated genome and large quantities of experimental data and publications are available.
Tip
This dataset can help you to complete your expert annotation.
How to read the result table?
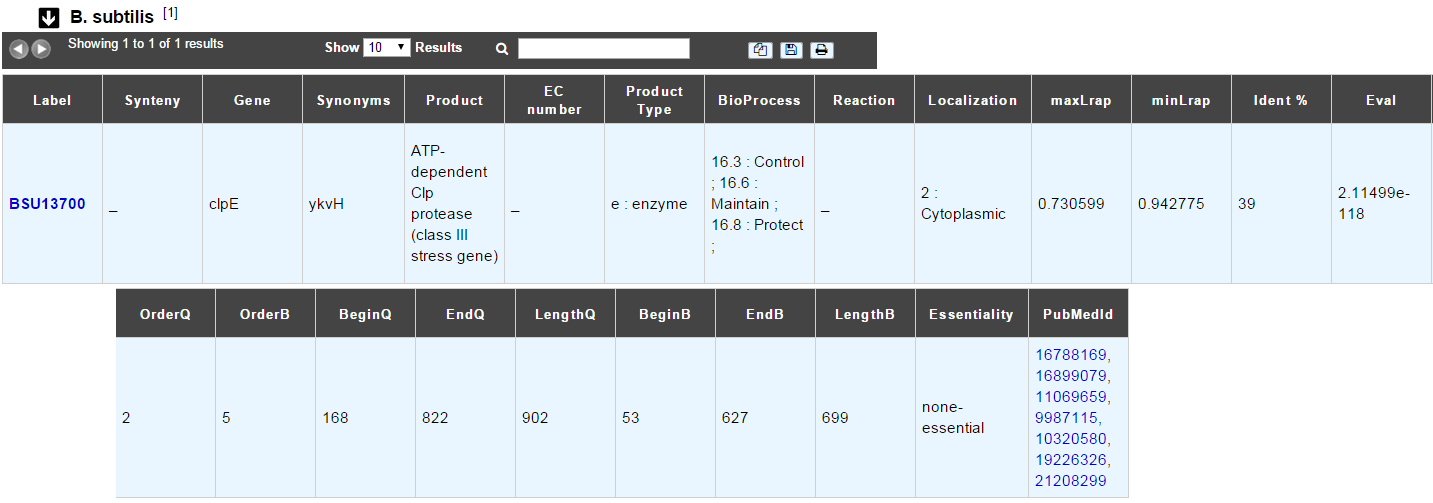
Label: Label of the protein. If you click on the label, you access to the Gene annotation window
Synteny: If you click on the magnifying glass, it opens a synton visualisation window (if any)
Gene: Gene name of the protein
Synonyms: Alternative name of the gene (if any)
Product: Product description of the protein
ECnumber: EC number associated with the protein, if any
Product type: Description of the product type of the protein
BioProcess: Functional categories associated with the protein using the BioProcess Functional classification
Reaction: If any, gives the reactions implying the database protein (reactions given by Rhea and MetaCyc)
Localization: Cellular localization of the protein
maxLrap: see BLAST results
minLrap: see BLAST results
Ident%: Percentage of identity between the studied protein and the database protein
Eval: E value of the BLAST result
OrderQ: see BLAST results
OrderB: see BLAST results
BeginQ: Start of the alignment for the studied protein
EndQ: End of the alignment for the studied protein
LengthQ: Length of the studied protein
BeginB: Start of the alignment for the database protein
EndB: End of the alignment for the database protein
LengthB: Length of the database protein
PubMedId: PubMed references linked to the annotation of the protein
DB of essential genes
This menu gives BLAST hits for the current Genomic Object against the essential gene database for genes with “essential” status.
This dataset comes from Database of Essential Genes (DEG) . DEG hosts records of currently available essential genomic elements, such as protein-coding genes and non-coding RNAs, among bacteria, archaea and eukaryotes. Essential genes in a bacterium constitute a minimal genome, forming a set of functional modules, which play key roles in the emerging field, synthetic biology. DEG database has been improved with data from Acinetobacter baylyi ADP1 and Neisseria meningitidis 8013, two highly curated genome in MicroScope.
How to read the result table?
Label: Label of the protein in DEG
Organism: reference organism in DEG
Gene: Gene name of the protein in DEG
PB id: Uniprot ID of the database protein. If you click on this Id, you can access the Uniprot profile of the protein, giving you various informations about it
Product: Product description of the protein in DEG
maxLrap: see BLAST results
minLrap: see BLAST results
Ident%: Percentage of identity between the studied protein and the database protein
Eval: E value of the BLAST result
OrderQ: see BLAST results
OrderB: see BLAST results
Exp condition: Experimental condition for essential characterization
PubMedId: PubMed references linked to the annotation of the protein
Source: Source of the reference data (DEG or MicroScope)
BeginQ: Start of the alignment for the studied protein
EndQ: End of the alignment for the studied protein
LengthQ: Length of the studied protein
BeginB: Start of the alignment for the database protein
EndB: End of the alignment for the database protein
LengthB: Length of the database protein
Favourite Genomes
This section indicates the best BLAST hits for the current Genomic Object with Genomic Objects from your favourite genome cart.
These other Genomic Objects having been automatically (re-)annotated using the MaGe platform, and maybe even been manually annotated/curated by MaGe users, can serve as informative references for your own annotations.
How to read the result table?
Label: Label of the protein. If you click on the label, you access the Gene annotation window for that Genomic Object.
Organism: Organism name. If you click on the name, you access the organism’s sequences on the NCBI website
Gene: Gene name of the protein
Evidence: Status of the annotation.
Gene: Gene name of the genomic object
Product: Product description of the protein
maxLrap: see BLAST results
minLrap: see BLAST results
Ident%: Percentage of identity between the studied protein and the database protein
Eval: E value of the BLAST result
OrderQ: see BLAST results
OrderB : see BLAST results
BeginQ: Start of the alignment for the studied protein
EndQ: End of the alignment for the studied protein
LengthQ: Length of the studied protein
BeginB: Start of the alignment for the database protein
EndB: End of the alignment for the database protein
LengthB: Length of the database protein
MaGe/Curated annotations
This section indicates the best BLAST hits obtained with other Genomic Objects from PkGDB which have been manually annotated/curated by other MaGe users.
How to read the result table?
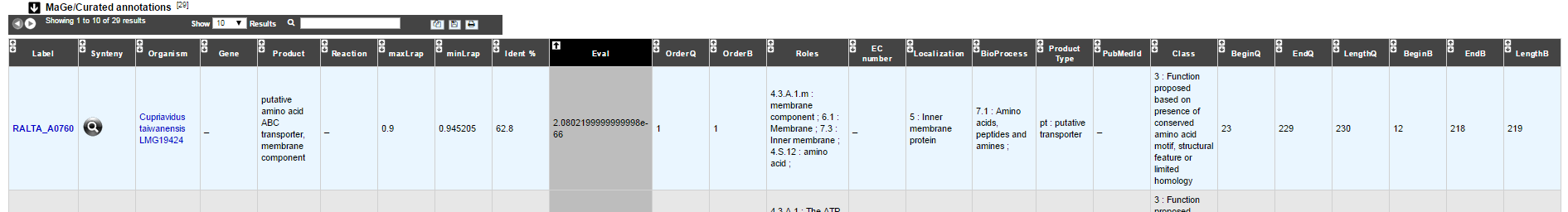
Label: Label of the protein. If you click on the label, you access to the Gene annotation window
Synteny: If you click on the magnifying glass, it opens a synton visualisation window
Organism: Organism name. If you click on the name, you access to the sequences on the NCBI website
Gene: Gene name of the protein
Product: Product description of the protein
maxLrap: see BLAST results
minLrap: see BLAST results
Ident%: Percentage of identity between the studied protein and the database protein
Eval: E value of the BLAST result
OrderQ: see BLAST results
OrderB: see BLAST results
Roles: Functional categories associated with the protein using the Roles functional classification
ECnumber: EC number associated with the protein, if any
Localization: Cellular localization of the protein
BioProcess: Functional categories associated with the protein using the BioProcess functional classification
Product type: Description of the product type of the protein
PubMedId: PubMed references linked to the annotation of the protein
Class: Confidence class of the annotation
BeginQ: Start of the alignment for the studied protein
EndQ: End of the alignment for the studied protein
LengthQ: Length of the studied protein
BeginB: Start of the alignment for the database protein
EndB: End of the alignment for the database protein
LengthB: Length of the database protein
Syntonome / Syntonome RefSeq
How to use the Syntonome / Syntonome RefSeq results?
These sections give access to the list of syntons which contain homologs to the studied gene in other organisms:
from PkGDB for the Syntonome section
from RefSeq for the Syntonome RefSeq section
How to read Syntonome results?
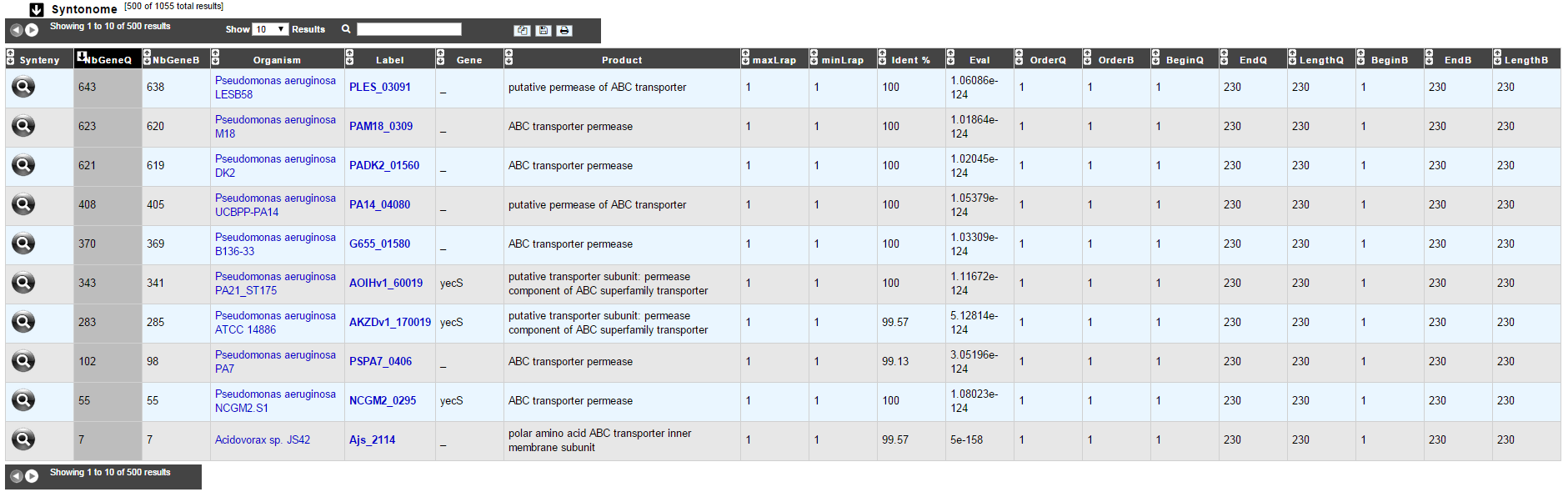
Synteny: If you click on the magnifying glass, it opens a synton visualisation window
NbGeneQ: Number of genes involved in the synton in the studied genome
NbGeneB: Number of genes involved in the synton in the database genome
Organism: Organism name. If you click on the name, you can access the associated genome sequence on the NCBI website.
Label: Label of the database protein. If you click on the label, you can access the Gene annotation window (Syntonome) or to the corresponding NCBI entry (Syntonome RefSeq)
Gene: Gene name of the database protein
Product: Product description of the database protein
maxLrap: see BLAST results
minLrap: see BLAST results
ident%: Percentage of identity between the studied protein and the database protein
Eval: E value of the BLAST result
OrderQ: see BLAST results
OrderB: see BLAST results
BeginQ: Start of the alignment for the studied protein
EndQ: End of the alignment for the studied protein
LengthQ: Length of the studied protein
BeginB: Start of the alignment for the protein of the database
EndB: End of the alignment for the protein of the database
LengthB: Length of the protein of the database
Similarities SwissProt / TrEMBL
What is UniProt?
The Universal Protein Resource (UniProt) is a comprehensive resource for protein sequence and annotation data. The mission of UniProt is to provide the scientific community with a comprehensive, high-quality and freely accessible ressource of protein sequence and functional information.
The UniProt Knowledgebase consists of two sections:
Swiss-Prot which contains high quality manually annotated and non-redundant protein sequences. This database brings together experimental results, computed features and scientific conclusions.
TrEMBL which contains protein sequences associated with computationally generated annotation and large-scale functional characterization that await full manual annotation.
More than 99% of the protein sequences provided by UniProtKB are derived from the translation of the coding sequences (CDS) which have been submitted to the public nucleic acid databases, the EMBL-Bank/GenBank/DDBJ databases. All these sequences, as well as the related data submitted by the authors, are automatically integrated into UniProtKB/TrEMBL.
More: http://www.uniprot.org/
How to read SwissProt and TrEMBL results?

PB id: Uniprot ID of the database protein. If you click on this Id, you can access the Uniprot profile of the protein, giving you various informations about it.
Exp: Indicates if there is PubMed references for the database protein. If there is at least one article, the mention “IPMed?” is written in this column.
maxLrap: see BLAST results
minLrap: see BLAST results
ident%: Percentage of identity between the studied protein and the database protein
Eval: E value of the BLAST result
OrderQ: see BLAST results
OrderB: see BLAST results
Gene: Gene name of the database protein
Description: Product description of the database protein
EC Number: gives the EC number (if any)
Keywords: Keywords associated to the protein function and roles
PubMedId: References linked to the annotation of the protein
Organism: Organism name. If you click on the name, you can access the associated genome sequence on the NCBI website.
Strain: Strain where the gene of the database is localized
BeginQ: Start of the alignment for the studied protein
EndQ: End of the alignment for the studied protein
LengthQ: Length of the studied protein
BeginB: Start of the alignment for the protein of the database
EndB: End of the alignment for the protein of the database
LengthB: Length of the protein of the database
UniFIRE
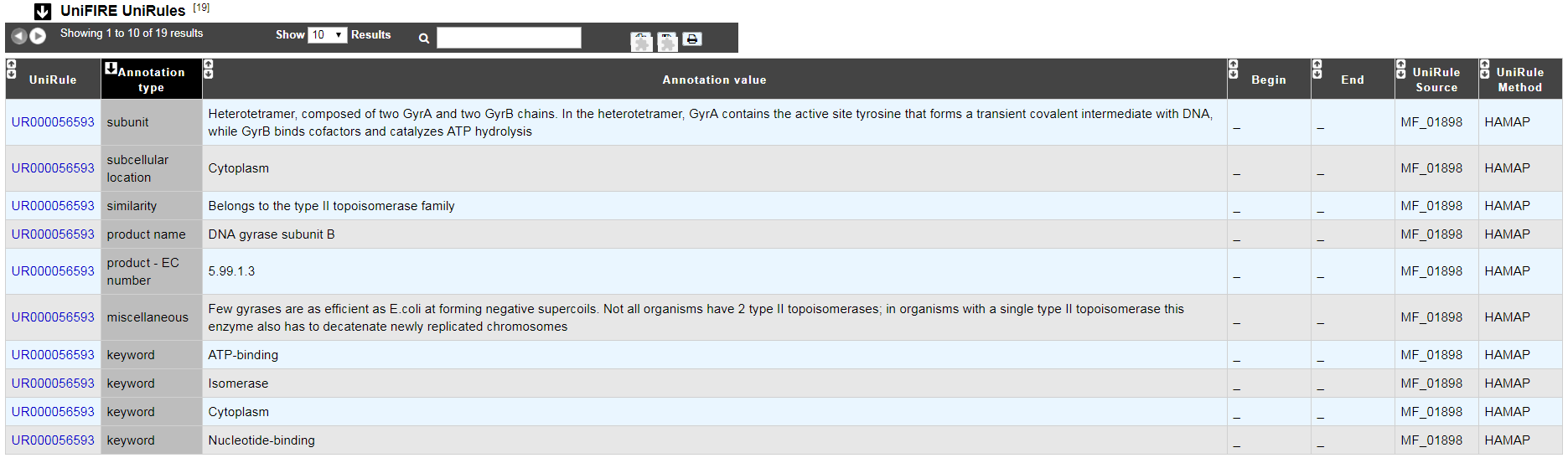
What is the UniFIRE?
UniFire (the UNIprot Functional annotation Inference Rule Engine) is a tool to apply the UniProt annotation rules. Two set of rule are applied :
The SAAS rules (Statistical Automatic Annotation System). This rules is generated automatic from expertly annotated entries in UniProtKB/Swiss-Prot.(https://www.uniprot.org/help/saas)
The UniRules (The Unified Rule) are devised and tested by experienced curators using experimental data from manually annotated entries.(https://www.uniprot.org/help/unirule)
How to read UniFIRE results?
UniRule : Rule id
Annotation type : Prediction type inferred
Annotation value : Annotation inferred
Begin : Start position of the predicted features
End : Enf position of the predicted features
UniRule Source : Source rule id
UniRule Method : Source rule
KOfamScan
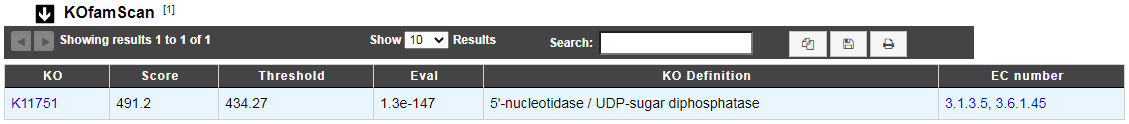
What is KOfamScan?
The KO KEGG Orthology is a database of molecular functions represented in terms of functional orthologs. The KO assignment of a gene allows to predict a function and an associated biological process.
For KO assignement, we use KOfamScan, a tool to homology search against a database of profile hidden Markov models (KOfam) with pre-computed adaptive score thresholds. In MicroScope, only the results that match with a score exceeding the pre-computed threshold and a E-value lower than or equal to 0.01 are shown.
Reference:
How to read KOfamScan results ?
KO : ID number of KEGG Ortholog entry
Score : Score of the match
Threshold : Pre-computed adaptive score for each KOfam
Eval : E-value of the match
KO Definition : Functional description of KO
EC number : EC number associated with the KO
Predicted MetaCyc Pathways
What are MetaCyc Pathways?
MetaCyc pathways are metabolic networks as define in the MetaCyc Database.
The presence or absence of a MetaCyc metabolic pathway is predicted by the Pathway-tools algorithm in this organism.
P. Karp, S. Paley, and P. Romero “The Pathway Tools Software,” Bioinformatics 18:S225-32 2002
How to read MetaCyc results?
All pathways listed in this table are those predicted as present in this organism. Clicking on the name of a pathway opens its table of reactions content.

EGGNOG
What is EGGNOG?
eggNOG (evolutionary genealogy of genes: Non-supervised Orthologous Groups) is a public resource in which thousands of genomes are analyzed at once to establish orthology relationships between all their genes.
An Orthologous Group (OG) is defined as a cluster of three or more homologous sequences that diverge from the same speciation event.
More information about the method: http://eggnogdb.embl.de/#/app/methods
eggNOG-mapper (version 2.1.12) is a tool for fast functional annotation of novel sequences. It uses precomputed orthologous groups and phylogenies from the eggNOG database to transfer functional information We run eggnog-mapper using EGGNOGDB and diamond for the alignement.
More information about eggNOG-mapper: http://eggnog-mapper.embl.de/
How to read eggNOG results?
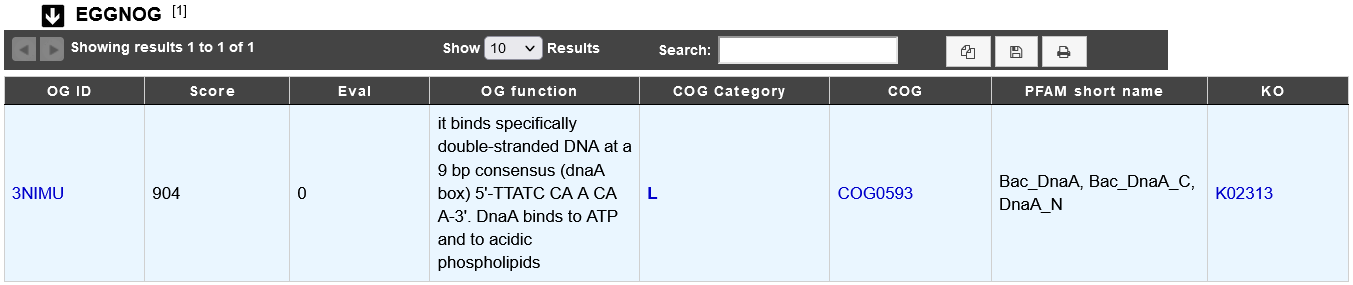
OG ID: eggNOG Orthologous Group id.
Score: score of the match.
Eval: E-value of the match.
OG function: eggNOG Orthologous Group functional description.
COG Category: COG functional category ID.
COG: Clusters of Orthologous Group name (COG).
PFAM short name: InterPro PFAM entry name.
KO: KEGG Ortholog entry ID.
FigFam
In progress
What is FigFam?
“FIGfams, a new collection of over 100 000 protein families that are the product of manual curation and close strain comparison. Using the Subsystem approach the manual curation is carried out, ensuring a previously unattained degree of throughput and consistency. FIGfams are based on over 950 000 manually annotated proteins and across many hundred Bacteria and Archaea. Associated with each FIGfam is a two-tiered, rapid, accurate decision procedure to determine family membership for new proteins. FIGfams are freely available under an open source license.” (quote from http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2777423/ )
How to read FigFam results?
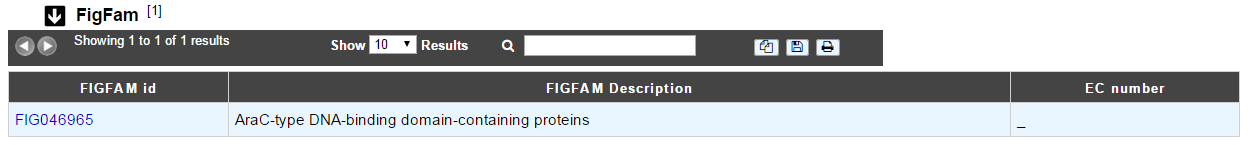
FIGFAM id: ID number of the FigFam family the protein is part of
FIGFAM Description: gives the description of the product of the family
EC number: gives the EC number
PsortB
What is PsortB?
PsortB is an open-source tool for protein sub-cellular localization prediction in bacteria.
More: http://www.psort.org/
How to read PsortB results?

The first column indicates the Localization predicted by PsortB.
The second column gives the score. The score typically varies between 2 and 10.
The third column indicates which option has been used for the genome: Gram positive (+) or Gram negative(-) bacteria.
InterProScan
What is InterPro?
InterPro is an integrated database of predictive protein “signatures” used for the classification and automatic annotation of proteins and genomes. InterPro classifies sequences at superfamily, family and subfamily levels, predicting the occurrence of functional domains, repeats and important sites. InterPro adds in-depth annotation, including GO terms, to the protein signatures.
Which databases are used in InterPro?
InterPro combines a number of databases (referred to as member databases) that use different methodologies and a varying degree of biological information on well-characterised proteins to derive protein signatures. By uniting the member databases, InterPro capitalises on their individual strengths, producing a powerful integrated database and diagnostic tool (InterProScan).
The member databases use a number of approaches:
PRODOM: provider of sequence-clusters built from UniProtKB using PSI-BLAST.
PROSITE (PROSITE patterns): provider of simple regular expressions.
PRINTS provider of fingerprints, which are groups of aligned, un-weighted Position Specific Sequence Matrices (PSSMs).
PANTHER, PIRSF, PFAM, SMART, TIGRFAMs, GENE3D and SSF (SUPERFAMILY): providers of hidden Markov models (HMMs).
CDD Conserved Domains and Protein Classification
SFLD A hierarchical classification of enzymes that relates specific sequence-structure features to specific chemical capabilities
Diagnostically, these resources have different areas of optimum application owing to the different underlying analysis methods. In terms of family coverage, the protein signature databases are similar in size but differ in content. While all of the methods share a common interest in protein sequence classification, some focus on divergent domains (e.g., Pfam), some focus on functional sites (e.g., PROSITE), and others focus on families, specialising in hierarchical definitions from superfamily down to subfamily levels in order to pin-point specific functions (e.g., PRINTS). TIGRFAMs focus on building HMMs for functionally equivalent proteins and PIRSF always produces HMMs over the full length of a protein and have protein length restrictions to gather family members. HAMAP profiles are manually created by expert curators they identify proteins that are part of well-conserved bacterial, archaeal and plastid-encoded proteins families or subfamilies. PANTHER build HMMs based on the divergence of function within families. SUPERFAMILY and Gene3D are based on structure using the SCOP and CATH superfamilies, respectively, as a basis for building HMMs.
How to read InterProScan results?
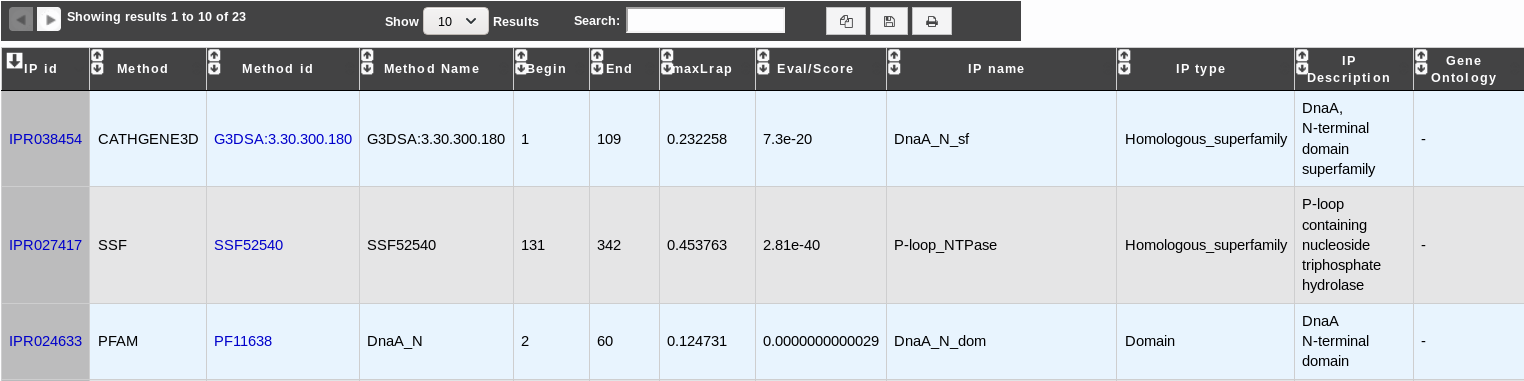
IP id: Identifier of the InterPro entry. Click on it to access the full description of the InterPro entry.
Method: Method used to obtain the result. It corresponds to one of the member database methods of InterPro.
Method id: Identifier of the method entry that generated the result. Click on it to access the full description of the method entry.
Method Name: Name of the method entry.
Begin: Beginning of the match on the query sequence.
End: End of the match on the query sequence.
maxLrap: Alignment coverage on the query sequence. See BLAST results.
Eval/Score: E-value or score of the match (if applicable).
IP name: Name of the InterPro entry.
IP type: Type of the InterPro entry.
IP description: Description of the InterPro entry.
Gene Ontology: Gene Ontology terms associated with the InterPro entry.
SignalP
What is SignalP?
SignalP (version 4.1) predicts the presence and location of signal peptide cleavage sites in amino acid sequences from different organisms: Gram-positive prokaryotes, Gram-negative prokaryotes, and eukaryotes. The method incorporates a prediction of cleavage sites and a signal peptide/non-signal peptide prediction based on a combination of several artificial neural networks and hidden Markov models.
Reference:
How to read SignalP results?
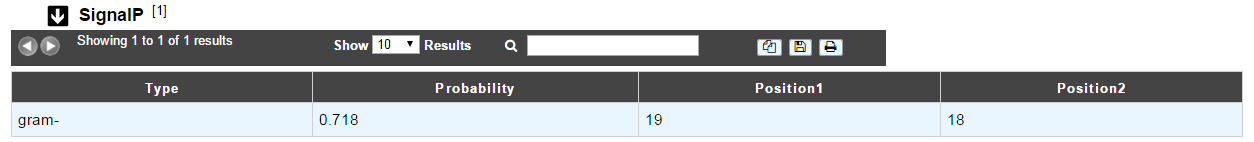
The first column indicates the type of bacteria (Gram positive or Gram negative).
The second column gives the estimated probability (number between 0 and 1) that the sequence contains a signal peptide.
The last 2 columns indicate the positions between which the cleavage is supposed to occur.
Tip
A signal peptide has an average size of 30 aa.
TMHMM
What is TMHMM?
TMHMM (version 2.0c) is a program for the prediction of transmembrane helices based on a hidden Markov model. The program reads a fasta-formatted protein sequence and predicts locations of transmembrane, intracellular and extracellular regions.
More: http://www.cbs.dtu.dk/services/TMHMM/
References:
How to read TMHMM results?
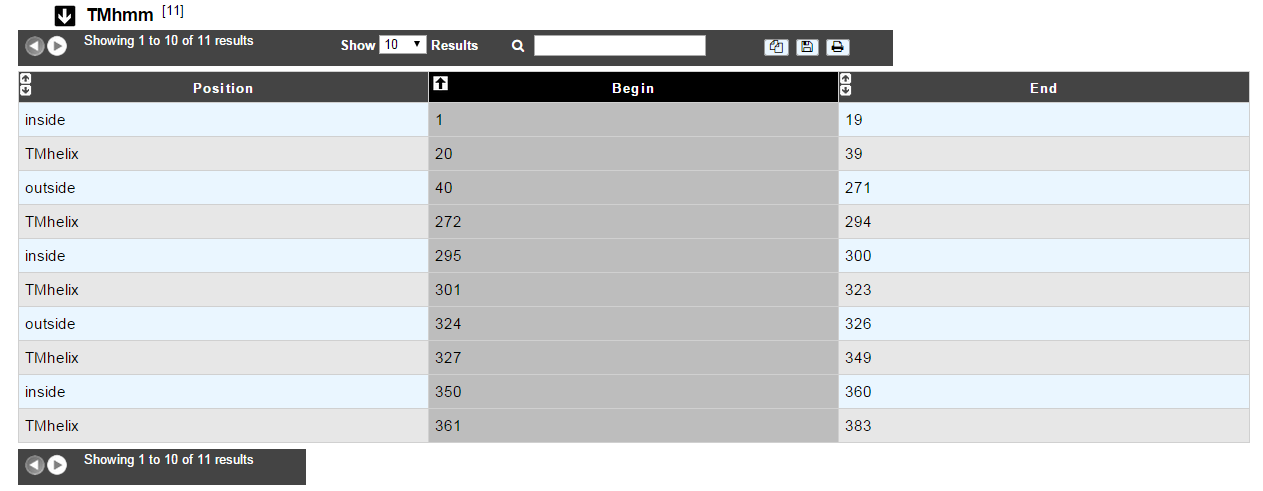
The table of results indicates the begin and end positions of detected alpha-helices for the protein sequence. It also gives the location (inside/outside) of the fragments in between the helices.
Tip
A protein can be called « membranar » if it contains more than 3 alpha-helices.
AntiSMASH
What is antiSMASH?
antiSMASH allows the rapid genome-wide identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genomes. It integrates and cross-links with a large number of in silico secondary metabolite analysis tools that have been published earlier.
More: http://antismash.secondarymetabolites.org/#!/about
Reference:
What type of secondary metabolites can antiSMASH 7.1.0.1 predict?
The list of all secondary metabolites predicted by antiSMASH is available in the glossary section of the antiSMASH documentation.
How to read antiSMASH 7.1.0.1 results?
AntiSMASH results are presented into 2 separate datasets: antiSMASH annotation and antiSMASH domains.
The antiSMASH annotation dataset:

antiSMASH Region id: antiSMASH region number. By clicking on the number, you can access to the antiSMASH cluster visualisation window.
Status: Gene status proposed by the tool (biosynthectic-additional, biosynthetic, transport, regulatory, resistance, other).
antiSMASH annotation: Predicted domains and/or SMCOG annotation, if any.
The antiSMASH domains dataset:

Sequence: If you click on the icon, it opens a window with the nucleic and proteic sequences of the domain.
Domain type: Domain type.
Begin and End: Location of the domain on the sequence.
Score: BLAST score.
E-value: BLAST E-value.
Substrate specificity prediction: Substrate specificity prediction of the domain predicted by antiSMASH, if any.
How can I visualize the clusters predicted by antiSMASH?
You can access to the antiSMASH cluster visualisation window by clicking on the number indicated in the antiSMASH Region id field of the antiSMASH annotation table. This window allows you to visualize the full antiSMASH cluster prediction and its genomic context.
LipoP
What is LipoP?
LipoP is a method to predict lipoprotein signal peptide. It is based on Hidden Markov Model (HMM) which discriminate lipoproteins (SPaseII-cleaved proteins), SPaseI-cleaved proteins, cytoplasmic proteins and transmembrane proteins. Although LipoP1.0 has been trained on sequences from Gram-negative bacteria only, the following paper (Methods for the bioinformatic identification of bacterial lipoproteins encoded in the genomes of Gram-positive bacteria; O. Rahman, S. P. Cummings, D. J. Harrington and I. C. Sutcliffe; World Journal of Microbiology and Biotechnology 24(11):2377-2382 (2008)) reports that it has good performance on sequences from Gram-positive bacteria also.
References:
How to read LipoP results?
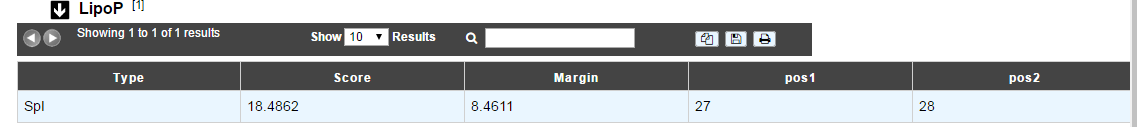
Type: type of the signal peptide (SPI or SPII)
Score: detection score
Margin: difference between the best and the second best score.
Pos1 and Pos2 indicate the positions between which the cleavage is supposed to occur
dbCAN
What is dbCAN?
dbCAN3 is a method for automated Carbohydrate-active enzyme ANnotation (dbCAN) and substrate annotation. The method incorporates HMMER, diamond, and dbCAN-sub for annotating CAZyme families and for substrate predictions. [1]
Carbohydrate-Active enZyme (CAZymes) are classified in the CAZy database which describes the families of structurally-related catalytic and carbohydrate-binding modules (or functional domains) of enzymes that degrade, modify, or create glycosidic bonds. [2]
dbCAN-sub is a profile Hidden Markov Model database (HMMdb) for substrate prediction at the CAZyme subfamily level. Each dbCAN-sub HMM corresponds to an eCAMI subfamily, containing protein sequences annotated by CAZy, some of which may have been experimentally characterized with EC numbers. They enable substrate prediction and provides subfamily-level annotations enriched with EC numbers and subfamily composition. In the following, they are named dbCAN-sub subfamilies but they can also be referred to as eCAMI subfamilies or even CAZyme subfamilies. They are listed on the dbCAN-sub website. [3]
For automated CAZyme annotation, the standalone version of the dbCAN3 known as run_dbcan conducts 3 tool–database annotation steps :
DIAMOND against CAZy compares sequences against
CAZyDB(the library of sequences classified into CAZy families) using diamond.HMMER against dbCAN compares sequences against
dbCAN HMMdb(the library of CAZy families HMM profiles) using HMMER.HMMER against dbCAN-sub compares sequences against
dbCAN-sub HMMdb(the library of dbCAN-sub subfamilies HMM profiles) using HMMER.
All databases used are downloaded by the tool run_dbcan when installed and can be found at that link.
References:
How to read dbCAN results?
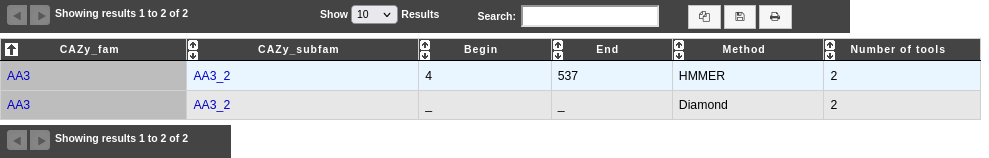
Each line presents a result of one genomic object by one of the tool–database annotation step used by dbCAN3 (one genomic object can have multiple results and each result is on its own line):
Method:
CAZy: DIAMOND = line displaying DIAMOND’s best match of the query sequence against
CAZyDB.dbCAN: HMMER = line displaying HMMER’s best match of the query sequence against
dbCAN HMMdb.dbCAN-sub: HMMER = line displaying HMMER’s best match of the query sequence against
dbCAN-sub HMMdb.
CAZy classification: Name of the predicted CAZy family with a link to the corresponding CAZy web page.
For CAZy: DIAMOND lines, it gives the family annotation of the matched protein sequence in
CAZyDB.For dbCAN: HMMER lines, it gives the family corresponding to the matched HMM profile in
dbCAN HMMdb.For dbCAN-sub: HMMER lines, it is the family obtained from truncating the dbCAN-sub Classification at the first
_(example: when dbCAN-sub Classification isCE11_e22, its CAZy Classification isCE11).
DIAMOND CAZyme: Protein accession of the best diamond match of the query sequence against
CAZyDB. It may appear in two formats:GenBank format: a unique accession identifier assigned to a protein sequence (e.g.,
QJW36450.1).JGI format: a sequence ID and the source fasta name (e.g.,
118117, Volvo1_GeneCatalog_proteins_20130703.aa.fasta).
DIAMOND EC number: Enzyme Commission number (EC number) describing the catalytic activity of the enzyme. Reported by DIAMOND when the matched sequence in
CAZyDBis annotated with an EC number.dbCAN-sub Classification: Subfamily name of the best HMMER match of the query sequence against
dbCAN-sub HMMdb, linked to the corresponding web page.dbCAN-sub Subfamily Composition: Composition in CAZy families of sequences composing the dbCAN-sub subfamily. Their number of occurence in the group is given next to the family.
Example:
GT4_e480 | GT4:76, GT2:6
Here, subfamily
GT4_e480is a cluster of sequences with 76 occurences ofGT4signature and 6 occurences ofGT2signature.
dbCAN-sub EC number: Enzyme Commission number (EC number) describing the catalytic activity of the enzyme. Predicted by dbCAN-sub when the assigned dbCAN-sub subfamily has a known functional annotation.
dbCAN-sub Substrate: Predicted substrate for each enzyme, inferred using the dbCAN-sub method.
Each substrate corresponds to an EC number of the enzyme subfamily.
Multiple substrates and EC numbers are listed in the same order, separated by “, “.
Example:
CAZy Classification | dbCAN-sub Classification | dbCAN-sub EC number | dbCAN-sub Substrate -------------------- | ------------------------ | --------------------- | ------------------- GH4 | GH4_e6 | 3.2.1.139, 3.2.1.20 | xylan, alpha-glucan
- Here:
xylanis associated with EC3.2.1.139andGH4.alpha-glucanis associated with EC3.2.1.20andGH4.
If the EC number is incomplete or missing, dbCAN-sub attempts to assign the most likely substrate based on the enzyme family.
Output preserves the EC order and shows one substrate or a group of substrates separated by “;” per EC.
If no match is found, the field will display nothing,
-orunknown.Refer to dbCAN publication for further information. [1]
Coverage: Fraction of the HMM profile covered by the alignment (reported for HMMER methods only; empty for DIAMOND results).
Ident%: Percentage of identical residues between the query and the subject sequence (reported for DIAMOND results only; empty for HMMER methods).
Eval: Expectation value of the alignment, reported by all 3 methods. Lower values indicate more significant matches.
BeginQ: Start position of the alignment on the query sequence.
EndQ: End position of the alignment on the query sequence.
LengthQ: Total length of the query sequence.
BeginB: Start position of the alignment on the subject (HMM profile or CAZy reference sequence).
EndB: End position of the alignment on the subject (HMM profile or CAZy reference sequence).
LengthB: Length of the subject sequence (HMM profile or CAZy reference).
GT2 special case
One may notice that in the dbCAN-sub Classification column, the family GT2 appears, eventhough the standard dbCAN-sub subfamily format includes “_e”. As clarified in the discussion of the related GitHub issue, GT2 is a special case in the dbCAN-sub pipeline.
As a result, several information normally provided by dbCAN-sub: HMMER cannot be delivered by the tool itself:
the specific
GT2subfamily assignmentthe composition of that subfamily
the associated EC number(s)
the predicted substrate
In short, this behavior is expected and intrinsic to how dbCAN-sub treats the GT2 family; the missing data is not an error and cannot be retrieved. Since the specific GT2 subfamily assignment is missing, it’s length is not be displayed.
SulfAtlas
What is SulfAtlas?
SulfAtlas is a tool for the automated detection of sulfatases. They are classified into the SulfAtlas database. From each SulfAtlas family and sub-family an HMM was generated. Thresholds have been manually defined.
- References:
Mark Stam, Pernelle Lelièvre, Mark Hoebeke, Erwan Corre, Tristan Barbeyron & Gurvan Michel, SulfAtlas, the sulfatase database: state of the art and new developments (manuscript in preparation)
How to read SulfAtlas results?
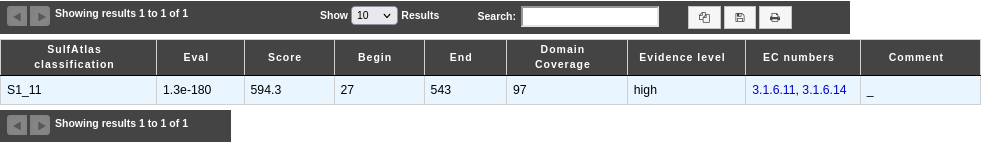
Each line presents a correspondence between a region of the genomic object and an HMM profile:
SulfAtlas classification: name of the SulfAtlas family/sub-family
Eval: e-value of the alignment
Score: score of the alignment
Begin: start position of the alignment on the protein sequence
End: end position of the alignment on the protein sequence
Domain Coverage: percentage of the sulfatase domain covered by the domain detected into the protein
Evidence level: give an level of evidence:
probable fragment means that the detected domain on the protein covers less than 80% of the sulfatase domain.
low means that the alignment score is under the threshold to assign the domain to a specific family/sub-family but high enough to consider that this domain is a sulfatase.
high means that the prediction is correct.
EC numbers: prediction of EC numbers, based on the EC numbers present into the SulfAtlas sub-family
Comment: Possible comment
Resistome
What is AMRFinderPlus?
The AMRFinderPlus software and the accompanying database are designed to find acquired antimicrobial resistance genes and point mutations in protein sequences.
AMRFinderPlus uses an AMR protein database, HMMs, a hierarchy of AMR protein families, and a custom rule set to identify AMR genes and point mutations, stress response genes, and virulence genes.
The database is highly curated with hierarchical structure for AMR proteins, manually curated cutoffs, and associated hierarchical names. It also includes point mutations, stress response and virulence genes, and it contains additional descriptive fields for each gene or point mutation.
References:
How to read AMRFinderPlus results?
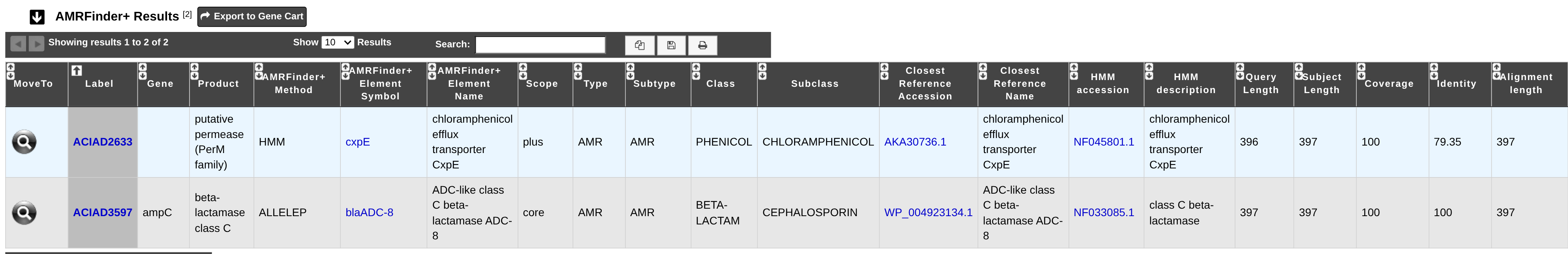
AMRFinderPlus is ran with the protein sequences as input using the ‘plus’ option and the ‘organism’ option.
the ‘plus’ option provides results from “Plus” genes such as virulence factors, stress-response genes, etc.
the ‘organism’ option delivers optimized organism-specific results (only for organisms which have been curated). It screens for point mutations, filter out non-informative genes and indentify specific features such as stx type in Escherichia or pbp proteins in Streptococcus pneumoniae or Neisseria gonorrhoeae.
All result fields are described in detail in the official AMRFinderPlus documentation. We recommand checking this page for more details.
Method: Type of hit detected by AMRFinderPlus (ALLELEP, EXACTP, PARTIALP, PARTIAL_CONTIG_ENDP, BLASTP, HMM, POINTP). The “P” suffix denotes that the analysis was performed using protein sequences.
AMRFinderPlus Element Symbol: Gene or gene-family symbol for protein or nucleotide hit, with a link to its entry in the Reference Gene Catalog.
AMRFinderPlus Element Name: Full-text name for the protein, RNA, or point mutation.
Scope: The AMRFinderPlus database is split into ‘core’ AMR proteins that are expected to have an effect on resistance and ‘plus’ proteins of interest added with less stringent inclusion criteria. These may or may not be expected to have an effect on phenotype.
Type and Subtype: These fields describe the classification of the AMRFinderPlus gene. “Element type” is split into 3 categories, AMR, STRESS, or VIRULENCE. “Element subtype” is a duplicate of “Element type” unless a more specific category has been defined.
Class: For AMR genes this is the class of drugs that this gene is known to contribute to resistance of.
Subclass: If more specificity about drugs within the drug class is known it is elaborated here.
Closest Reference Accession: RefSeq accession for reference hit by BLAST if a blast alignment was detected otherwise NA. The link directs to the corresponding entry in NCBI.
Closest Reference Name: Full name assigned to the closest reference hit if a blast alignment was detected, otherwise NA.
HMM accession: Accession for the HMM, with a link to its entry in the Reference HMM Catalog.
HMM description: The family name associated with the HMM.
Query Length: The length of the query protein or gene.
Subject Length: The length of the Reference protein in the database if a blast alignment was detected.
Coverage: % of reference covered by blast hit if a blast alignment was detected.
Identity: % amino-acid identity to reference protein if a blast alignment was detected.
Alignment length: Length of BLAST alignment in amino-acids if a blast alignment was detected.
You can access AMRFinderPlus predictions via the Resistome page, located in the Comparative Genomics section of the main navigation menu.
Virulome
What is VirulenceDB?
VirulenceDB is a virulence genes database build using three sets of data:
The core dataset from VFDB (setA), which is composed of genes associated with experimentally verified virulence factors (VFs) for 53 bacterial species
The VirulenceFinder dataset which includes virulence genes for Listeria, Staphylococcus aureus, Escherichia coli/Shigella and Enterococcus
A manually curated dataset of reference virulence genes for Escherichia coli (Coli_Ref).
The original virulence factors classification from VFDB has been hierarchically attributed to each gene as frequently as possible, in order to provide a functional interpretation of your results. New virulence factors have also been added to VirulenceFinder and Coli_Ref database to describe as best as possible the gene functions.
Know more about VFDB
Know more about VirulenceFinder
References:
How to read Virulome results?
Label / Gene / Product : Label, name of the gene and its product predicted by the Microscope platform
Virulence gene description : Vir Organism, Vir Gene, VF name, VF classes, VF pathotypes, VF structure, VF function, VF characteristic, VF mechanism
Result interpretation: Score from Blast, E-value, orderQ (rank of the BLAST hit for the protein of the query genome) and orderB (rank of the BLAST hit for the protein of the virulence database).
Additional information on VF classes:
They are divided into 4 main classes as proposed by VFDB:
Offensive virulence factors
Defensive virulence factors
Nonspecific virulence factors
Regulation of virulence-associated genes
A gene can be involved in many classes. For example, the gene kpsE (Capsule polysaccharide export inner-membrane protein KpsE) from E. coli can act both as an offensive virulence factor and a defensive virulence factor.
So the VF classes corresponding is “Offensive virulence factors, Invasion, Defensive virulence factors, Antiphagocytosis“ which correspond to :
Offensive virulence factors
1.1 Invasion
Defensive virulence factors
2.1 Antiphagocytosis
You can access to the Virulence Result page by clicking on Virulome tab in the Comparative Genomics menu.
IntegronFinder
What is IntegronFinder ?
IntegronFinder is a tool that detects integrons in DNA sequences with high accuracy. It combines the use of HMM profiles for the detection of integron integrases, and Covariance Models for the detection of attC sites.
IntegronFinder distinguishes 3 types of elements:
Complete integron: Integron including an integrase and at least one attC site,
In0 element: Integron including only an integrase,
CALIN (Clusters of AttC sites Lacking integron-INtegrases) element: Integron composed of at least two attC sites.
Know more about IntegronFinder.
Reference:
How to read IntegronFinder results ?
The IntegronFinder dataset appears if the genomic object belongs to an integron (e.g. antibiotic resistance gene).
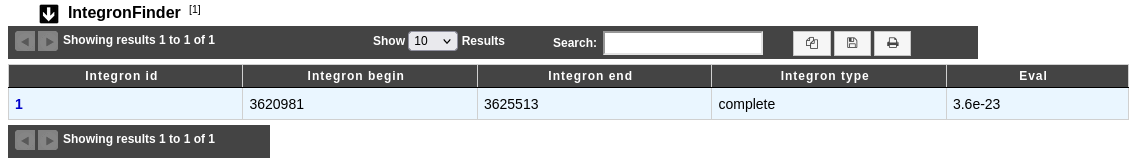
Integron id: Identifier of the integron where the integrase is found. Click on it open the detailled IntegronFinder cluster visualization window page, which allows to access to the description of the integron structure.
Integron begin and Integron end: Location of the integron on the sequence.
Integron type: Integron type (complete, In0, CALIN).
Eval: Evalue of the match with the integrase HMM profil.
MacSyFinder
What is MacSyFinder?
Macromolecular System Finder (MacSyFinder) provides a flexible framework to model the properties of molecular systems (cellular machinery or metabolic pathway) including their components, evolutionary associations with other systems and genetic architecture. Modelled features also include functional analogs, and the multiple uses of a same component by different systems. Models are used to search for molecular systems in complete genomes or in unstructured data like metagenomes. The components of the systems are searched by sequence similarity using Hidden Markov model (HMM) protein profiles. The assignment of hits to a given system is decided based on compliance with the content and organization of the system model.
Know more about MacSyFinder.
Reference:
How to read MacSyFinder results?
The MacSyFinder dataset appears if the genomic object corresponds to a macromolecular system predicted by MacSyFinder.

Macromolecular system: Label of the system predicted by MacSyFinder. Click on it open the MacSyFinder System visualization window page, which allows to access to a detailled description of the selected macromolecular system.
Mandatory genes present: Names of distinct mandatory genes composing the system.
Begin and End: Location of the system on the sequence.
Gene status: Status of the gene in the system (mandatory, accessory, neutral).
Hit label: Name of the MacSyFinder HMM profile which matchs with the sequence.
Eval: Evalue of the match.
Query coverage: Coverage of the match on the sequence.
Subject coverage: Coverage of the match on the MacSyFinder HMM profile.
Begin match and End match: Location of the match on the sequence.
Phigaro
What is Phigaro?
Phigaro is a standalone command-line application that is able to detect prophage regions taking raw genome and metagenome assemblies as an input. It also produces dynamic annotated “prophage genome maps” and marks possible transposon insertion spots inside prophages. It is applicable for mining prophage regions from large metagenomic datasets. Phigaro uses the pVOG HMM profiles to detect bacteriophage genes.
Know more about Phigaro.
Reference:
Note
By default Phigaro predicts genes by using Prodigal. However we use the gene calling provided by our own pipeline.
How to read Phigaro results?
The Phigaro dataset appears if the genomic object correspond to a prophage predicted by Phigaro The table shows :
System id: Id number of the macromolecular system to which belongs the gene
Prophage id: Id number of the prophage to which belongs the gene; clicking on this opens the prophage visualization interface
pVOG: the pVOG corresponding to the genomic object (if any); clicking on this will open the detailed description of the pVOG
Eval: E-value of the match between the genomic object and the pVOG

DefenseFinder
What is Defense Finder?
DefenseFinder is a program to systematically detect known anti-phage systems based on MacSyFinder. The decision rules are typically defined by a list of mandatory, accessory, or forbidden proteins necessary for the detection of a given system.
Know more about DefenseFinder.
- Reference:
How to read DefenseFinder results?
The DefenseFinder dataset appears if the genomic object corresponds to a defense system predicted by DefenseFinder.

Activity: Biological role of the system: “Defense” corresponds to anti-phage defense systems, whereas “Antidefense” refers to mechanisms that counteract prokaryotic defense systems.
System name: Name of the defense system to which belongs the gene, clicking on this opens the defense system visualization interface.
Protein name: Name of the protein detected.
Status: Status of the gene in the system (mandatory, accessory, neutral).
Eval: E-value of the match with DefenseFinder models.